Blodsyndrom: Symptomer, årsaker og behandling
Blodsyndrom (BS) er en sjelden sykdom av autosomal recessiv arv som hovedsakelig er preget av tre aspekter: vekstretardering, overfølsomhet overfor sol og telangiektasi i ansiktet (dilatasjon av kapillærkar). Disse pasientene har en genomisk ustabilitet som predisposes dem til å utvikle kreft enkelt.
Det ble oppdaget av dermatologen David Bloom i 1954 gjennom observasjon av flere pasienter som presenterte dwarfisme og telangiektatisk erytem (rødmet hud på grunn av utvidelse av blodkarillærer) (Elbendary, 2015).
Dette syndromet kan også kalles medfødt telangiectatisk erytem eller Bloom-Torre-Machacek syndrom.
Årsaker til Bloom syndrom
Bloom-syndromet er en autosomal resessiv sykdom, det vil si en mutasjon må forekomme i begge alleler av BLM-genet, både fra moren og fra faren (Ellis et al., 1995). Foreldre trenger ikke nødvendigvis å ha denne sykdommen, men de kan være bærere av det muterte genet uten å ha symptomer.
Mer enn 60 mutasjoner har blitt funnet i BLM-genet i Blooms syndrom, den hyppigste er sletting av 6 nukleotider i posisjon 2281 og substitusjonen av 7 andre (Elbendary, 2015).
Ifølge Genetics Home Reference (2016) er BLM-genet ansvarlig for å sende instruksjoner for opprettelsen av RecQ-proteinet, som er en del av familien av helikaser.
Hvilke helikaser gjør det er å slutte seg til DNA og midlertidig skille de to strengene av det, som vanligvis knyttes sammen i en spiral, med sikte på å utvikle prosesser som replikering (eller DNA-kopiering), forberedelse til celleoppdeling og reparasjon. av DNA-skade.
Kort sagt, RecQ helikaser er viktige for å opprettholde DNA-strukturen og er derfor kjent som "genomsorgere".
For eksempel, når en celle skal dele seg for å danne to nye celler, må DNA som er i kromosomene kopieres slik at hver ny celle har to kopier av hvert kromosom: en av far og en annen av moren.
DNA kopiert fra hvert kromosom har to identiske strukturer som kalles søsterkromatider, og de er koblet i begynnelsen, før delingen av cellene finner sted.
I dette stadiet bytter de noen deler av DNA mellom dem; hva er kjent som søster kromatid utveksling. Det ser ut som at denne prosessen er endret i Blooms sykdom, siden BLM-proteinet er skadet, og dette styrer de riktige byttene mellom søsterkromatidene og at DNA-en forblir stabil ved kopieringstidspunktet.
Faktisk er det gjennomsnittlig 10 mer enn normale utvekslinger mellom kromatider i Bloom syndromet (Seki et al., 2006).
På den annen side er det også brudd på det genetiske materialet i denne sykdommen, noe som fører til forringelse i normale cellulære aktiviteter som, på grunn av mangelen på BLM-proteinet, ikke kan repareres.
Faktisk klassifiserer noen eksperter dette syndromet som "kromosombruddssyndromet", siden det er relatert til et stort antall pauser og omorganiseringer av kromosomene.
Denne ustabiliteten av kromosomene forårsaker en større sannsynlighet for å utvikle sykdommer. For eksempel, på grunn av mangelen på BLM-proteinet, kan de ikke gjenopprette fra DNA-skade som kan forårsake ultrafiolett lys, og derfor er disse pasientene lysfølsomme.
I tillegg har de berørte en immunmangel som gjør dem mer utsatt for infeksjon.
På den annen side har de høy sannsynlighet for å utvikle kreft i et hvilket som helst organ ved den ukontrollerte delen av celler, som hovedsakelig forekommer leukemi (det er en type blodkreft som er preget av et overskudd av hvite blodlegemer) og lymfom (kreft i lymfeknuden til systemet). immun).
Feil har også blitt funnet i virkningen av FANCM-genet, som er ansvarlig for kodingen av MM1 og MM2-proteiner, som også tjener til å reparere DNA-skade.
Dette er de som har vært knyttet både til dette syndromet og til Fanconi anemi. Det er derfor vi ser at disse to sykdommene er like i deres fenotype og i predisponering mot hematologiske svulster og mangel i benmarg.
Uansett er molekylære mekanismer som påvirker kromosomene i Bloom syndromet fortsatt undersøkt.
Hva er dens prevalens?
Bloom-syndromet er relativt sjeldent, kun ca 300 tilfeller beskrevet i medisinsk litteratur er kjent. Selv om denne lidelsen forekommer hos mange etniske grupper, ser det ut til å være mye mer vanlig i Ashkenazi-jødene, og står for 25% av pasientene med dette syndromet.
Faktisk, innenfor denne etniske gruppen, kan frekvensen av å presentere syndromet nå 1%. Det har også blitt funnet, om enn mindre, i japanske familier.
Når det gjelder sex, synes menn å være litt mer sannsynlig å ha sykdommen enn kvinner, og andelen er 1, 3 menn for 1 kvinne.
Hva er dens symptomer?
Denne tilstanden oppstår allerede i de første månedene av livet, og for nå har ingen av pasientene levd mer enn 50 år.
- Ondartede svulster : forårsaket av genomisk ustabilitet som forklart ovenfor, er hovedårsaken til døden hos de som er rammet av dette syndromet. Ifølge National Organization for Rare Disorders (2014), vil ca 20% av de som rammes av Bloom syndrom utvikle kreft. Disse pasientene har mellom 150 og 300 ganger større risiko for å utvikle kreft enn mennesker uten denne lidelsen.
- Immundefekt som varierer i alvorlighetsgrad etter pasienten, og forutsettes for ulike infeksjoner. Dette skyldes underskudd i spredning av lymfocytter (hvite blodlegemer), problemer i syntesen av immunoglobulin (antistoffer i immunsystemet) og lav respons på stimulering av mitogener (som styrer divisjon og vekst av celler).
- Feil i T- og B-lymfocytter er hyppige og påvirker utviklingen av immunsystemet.
- Feilfunksjonen i immunsystemet kan føre til øreinfeksjon (hovedsakelig otitis media), lungebetennelse eller andre tegn som diaré og oppkast.
- Fotosensitivitet : Det er en overdreven følsomhet av DNA til ultrafiolette stråler, blir skadet. Det regnes som en form for fototoksisitet eller celledød som skader huden til de berørte når solen treffer.
- Reduksjon av fruktbarhet eller infertilitet . Faktisk er det hos menn ikke mulig å produsere venter. Hos kvinner er det en svært tidlig overgangsalder.
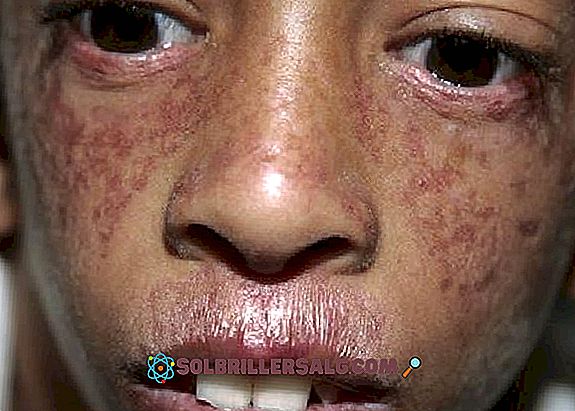
- Kutan manifestasjoner : I tillegg til lysfølsomhet opptrer poikiloderma også, en påvirkning av huden som forekommer hovedsakelig i nakken, som opptrer hypopigmenterte soner, andre hyperpigmenterte områder, telangiektasier og atrofi. Det er ofte observert røde flekker på huden som er forbundet med soleksponering (spesielt på ansiktet).
- Et annet kutan problem som observeres er telangiectasia, som ses som rødbrune utbrudd på ansiktet som skyldes utvidelse av små blodkar. Det ser ut som et "sommerfugl" mønster som dekker nesen og kinnene.
- Unormale brune eller grå flekker kan også vises på andre deler av kroppen ("café au lait" flekker).
- Forsinkelse i utviklingen som allerede manifesterer seg i babyer. De små har vanligvis et særegent hode og ansikt, smalere og mindre enn normalt.
- Om lag 10% av de som rammes, kommer til å utvikle diabetes (National Organization for Rare Disorders, 2014).
- Veldig skarp stemme .
- Endringer i tennene .
- Unormalitet i øynene, ørene (fremtredende ører), hender eller føtter (som polydaktyly, som oppstår når pasienten har flere fingre enn normalt).
- Pilonidale cyster.
- Feilproblemer : De blir lagt merke til spesielt hos spedbarn og små barn, og viser mangel på interesse for å spise. Det følger med mange ganger med alvorlig gastroøsofageal refluks.
- Den intellektuelle kapasiteten er variabel, slik at i noen pasienter blir de mer forverret og i andre er de innenfor normal.
Hvordan er det diagnostisert?
Det kan bli diagnostisert av noen av følgende tester:
- Cytogenetiske tester som måler kromosomavvik og nivået på utveksling av søsterkromatider.
Det er mulig å observere nærvær av fire-radiale foreninger (utveksling av firearmkromatider) i lymfocytter som er dyrket i blod, for å kontrollere om det er høye nivåer av utveksling av søsterkromatider i en hvilken som helst celle, kromatidgap, brudd eller omarrangementer; Eller se direkte om det er mutasjoner i BLM-genet.
Disse testene kan oppdage en sunn person som bærer mutasjoner i BLM-genet og kan overføre dem til sine avkom.
Food and Drug Administration of United States (FDA) kunngjorde i februar 2015 kommersialiseringen av en genetisk test av "23andMe" som kan være nyttig for å oppdage tidlig forekomsten av denne sykdommen.
Tilstedeværelsen av dette syndromet må antas hvis disse kliniske tilstandene eksisterer:
- Forsinkelse i den betydelige veksten sett siden intrauterin perioden.
- Tilstedeværelse av erytemmer på ansiktets hud etter eksponering for solen.
Ikke forveksle med ...
Følgende syndromer må tas i betraktning for å utelukke dem før de diagnostiserer Blooms syndrom:
- Andre autosomale recessive kromosomale ustabilitetssyndrom som er forbundet med brudd og omplasseringer av kromosomer, noe som gjør faget spesielt utsatt for visse typer kreft som: Fanconi anemi, ataxia telangiectasia eller xeroderma pigmentosa som involverer andre gener og ikke BLM .
- Cockayne syndrom, som består av en arvelig forstyrrelse manifestert av forsinket utvikling, lysfølsomhet og aldrende utseende i ung alder. Det er en sjelden form for dvergisme.
- Rothmund-Thomson syndrom : ekstremt uvanlig og manifestert av typiske hudabnormaliteter, hårfeil, juvenile katarakt, kort statur og skjelettendringer som kraniofacial misdannelser. Det ligner Blooms syndrom i hudbetennelse, poikiloderma, huddegenerasjon (atrofi) og telangiektasi.
behandling
Det er ingen spesifikk behandling for Bloom syndrom, det vil si for det store antallet mutasjoner. Snarere er inngrepene rettet mot å lindre symptomer, tilby støtte og forebygge komplikasjoner.
- Prøv å ikke utsette deg selv direkte under solen.
- Bruk en tilstrekkelig solkrem.
- Oppfølging av en hudlege, for å behandle flekker, rødhet og betennelse i huden.
- Bruk antibiotika for infeksjoner.
- Periodiske medisinske kontroller for å oppdage mulige tilfeller av kreft, hovedsakelig når disse pasientene når voksenalderen. Vi må forsøke å være oppmerksomme på de mulige symptomene, siden det er svulster som krever en tidlig kirurgisk fjerning for utvinning. Noen metoder for tidlig diagnose av kreft er mammografi, Papanicolaou eller vaginal cytologi, eller koloskopi.
- Kontroller at disse barna får de nødvendige næringsstoffene som prøver å gripe inn i fordøyelseskanalen. For dette kan et rør plasseres i den øvre delen av tarmkanalen for komplementær fôring mens du sover. Det kan øke de små fettavsetningene litt, men det ser ikke ut til å ha en effekt på selve veksten.
- Undersøk eksistensen av diabetes for å behandle den så snart som mulig.
- Hvis personen har kreft, kan beinmargstransplantasjon overveies.
- Familie og andre støttegrupper og foreninger med lignende sykdommer, slik at den berørte personen utvikler seg som en person, med høyest mulig livskvalitet.
- Hvis familien har hatt tilfeller av denne sykdommen eller av ektefelleens familie, ville genetisk rådgivning være nyttig for å få informasjon om natur, arv og konsekvenser av denne type lidelser for å bidra til medisinsk beslutningstaking og personlig.